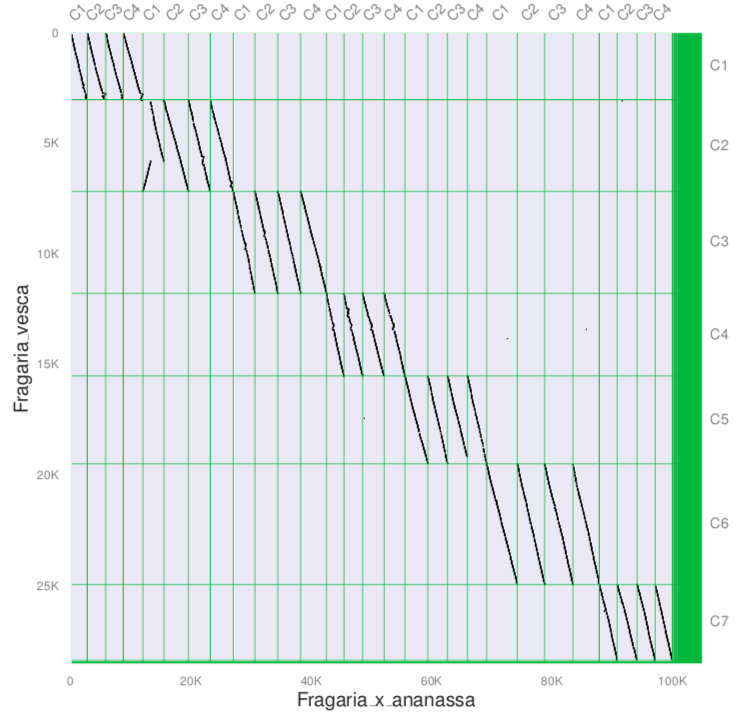
共线性(jcvi)
BioNote 2021-12-15
Collinearity
Comparative genomics
# 参考
# 文件准备
- gff文件
- cds文件或者蛋白文件 gff提供了基因的位置信息,cds或者蛋白用来进行比对,根据cds或者蛋白的比对结果进行位置上的锚定
# 软件信息
软件版本 : jcvi:v1.1.17-beta 安装方法见作者github
# 基因组共线性比对
# 转换gff文件
python3 -m jcvi.formats.gff bed --type=mRNA --key=ID unk.gff > unk.bed
1
type为gff格式第三列,代表去见对应的特征类型,key为该转录本的名称属性,该名称的值应该与cds或者蛋白名称对应。
if isoforms are excessive, we suggest adding one option --primary_only to the BED generation command above
# 序列比对
cds 比对
python3 -m jcvi.compara.catalog ortholog --no_strip_names unk unk2
1
或 蛋白比对
python3 -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names unk unk2
1
命名为unk.cds、unk.bed,jcvi会自动寻找目录下对应文件
如果想拿到1:1的同源区域,可以设置较高的C-score来获得best blast hit
rm unk.unk2.last.filtered
python3 -m jcvi.compara.catalog ortholog unk unk2 --cscore=.99 --no_strip_names
1
2
2
此时可以拿到一个点图和如下文件:
- unk.unk2.last: 基于LAST的比对结果
- unk.unk2.last.filtered: LAST的比对结果过滤串联重复和低分比对
- unk.unk2.anchors: 高质量的共线性区块
- unk.unk2.lifted.anchors:增加了额外的锚点,形成最终的共线性区块
每个共线性区块以###进行分隔, 第一列是检索的基因,第二列是被检索的基因,第三列则是两个序列的BLAST的score。
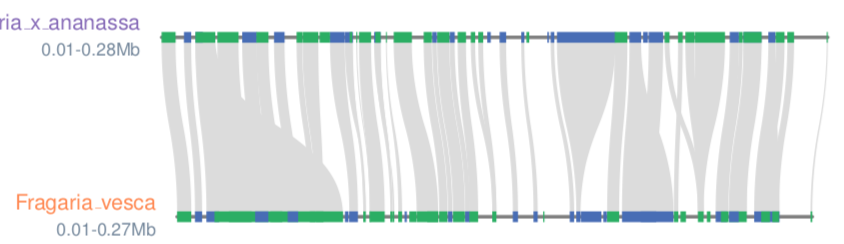
# 共线性绘图
根据anchors生成绘图所需的simple文件
python3 -m jcvi.compara.synteny screen --minspan=30 --simple unk.unk2.anchors unk.unk2.anchors.new
1
- seqids: 需要展现哪些序列
- layout: 不同物种的在图上的位置
- .simple: 从.anchors文件创建的简化格式,.simple文件中的每一行都是一个共线性块,物种1的基因起始终止,然后是物种2的基因起始终止,最后两列是 score 和orientation
seqids:
chr1-1,chr1-2,chr1-3,chr1-4,chr2-1,chr2-2,chr2-3,chr2-4,chr3-1,chr3-2,chr3-3,chr3-4,chr4-1,chr4-2,chr4-3,chr4-4,chr5-1,chr5-2,chr5-3,chr5-4,chr6-1,chr6-2,chr6-3,chr6-4,chr7-1,chr7-2,chr7-3,chr7-4
Fvb1,Fvb2,Fvb3,Fvb4,Fvb5,Fvb6,Fvb7
1
2
2
layout:
# y, xstart, xend, rotation, color, label, va, bed
.6, .2, .9, 0, , Fragaria_x_ananassa , top, Fragaria_x_ananassa.bed
.4, .4, .6, 0, , Fragaria_vesca , bottom, Fragaria_vesca.bed
# edges
e, 0, 1, Fragaria_x_ananassa.Fragaria_vesca.anchors.simple
1
2
3
4
5
2
3
4
5
最终绘图
python3 -m jcvi.graphics.karyotype seqids layout
1
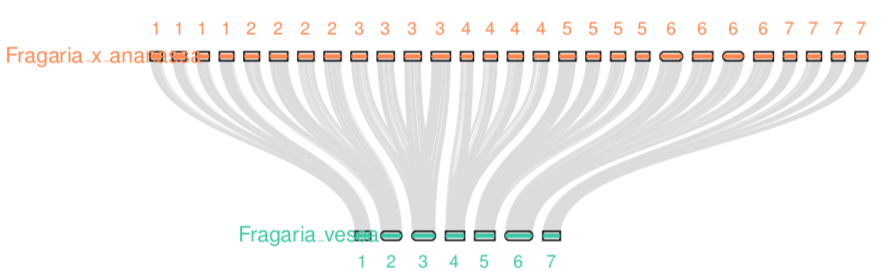
# 特定共线性区块着色
编辑.simple文件,.simple文件中每一行都是一个共线性区块,可以指定某个区块颜色(g*为绿色)
Fx0845400.1 Fx0832250.1 FvH4_1g00010.1 FvH4_1g15170.1 1412 +
g*Fx0832230.1 Fx0825230.1 FvH4_1g15420.1 FvH4_1g23510.1 753 +
Fx0820770.1 Fx0820240.1 FvH4_1g27990.1 FvH4_1g28580.1 56 -
Fx0820200.1 Fx0819070.1 FvH4_1g28600.1 FvH4_1g29420.1 97 -
g*Fx0819080.1 Fx0818340.1 FvH4_1g29790.1 FvH4_1g30420.1 69 +
Fx0727270.1 Fx0701540.1 FvH4_1g00010.1 FvH4_1g27480.1 2659 +
r*Fx0697480.1 Fx0696510.1 FvH4_1g27870.1 FvH4_1g28430.1 74 -
...
(save the file)
python3 -m jcvi.graphics.karyotype seqids layout
1
2
3
4
5
6
7
8
9
10
11
2
3
4
5
6
7
8
9
10
11
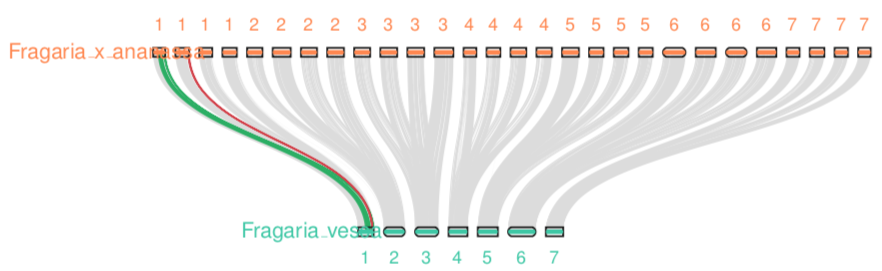
# 局部区域(gene-level)共线性
获取block文件,其中--iter代表有多少个match
python3 -m jcvi.compara.synteny mcscan Fragaria_vesca.bed Fragaria_x_ananassa.Fragaria_vesca.lifted.anchors --iter=1 -o Fragaria_x_ananassa.Fragaria_vesca.i1.blocks
1
选取需要绘制的block
head -n 50 Fragaria_x_ananassa.Fragaria_vesca.i1.blocks > blocks
1
编写布局文件blocks.layout
# x, y, rotation, ha, va, color, ratio, label
0.5, 0.6, 0, left, center, m, 1, Fragaria_x_ananassa
0.5, 0.4, 0, left, center, #fc8d62, 1, Fragaria_vesca
# edges
e, 0, 1
1
2
3
4
5
2
3
4
5
绘制基因水平共线性
cat Fragaria_x_ananassa.bed Fragaria_vesca.bed > Fragaria_x_ananassa.Fragaria_vesca.bed
python3 -m jcvi.graphics.synteny blocks Fragaria_x_ananassa.Fragaria_vesca.bed blocks.layout
1
2
2